





Overview
The Boettcher electrochemistry and materials science laboratory studies, designs, synthesizes, fabricates, and models materials and devices for applications in electrochemical energy storage and solar energy conversion. Our work spans use-inspired basic science to applied science and engineering, and technology transfer through industry collaborations and partnerships.
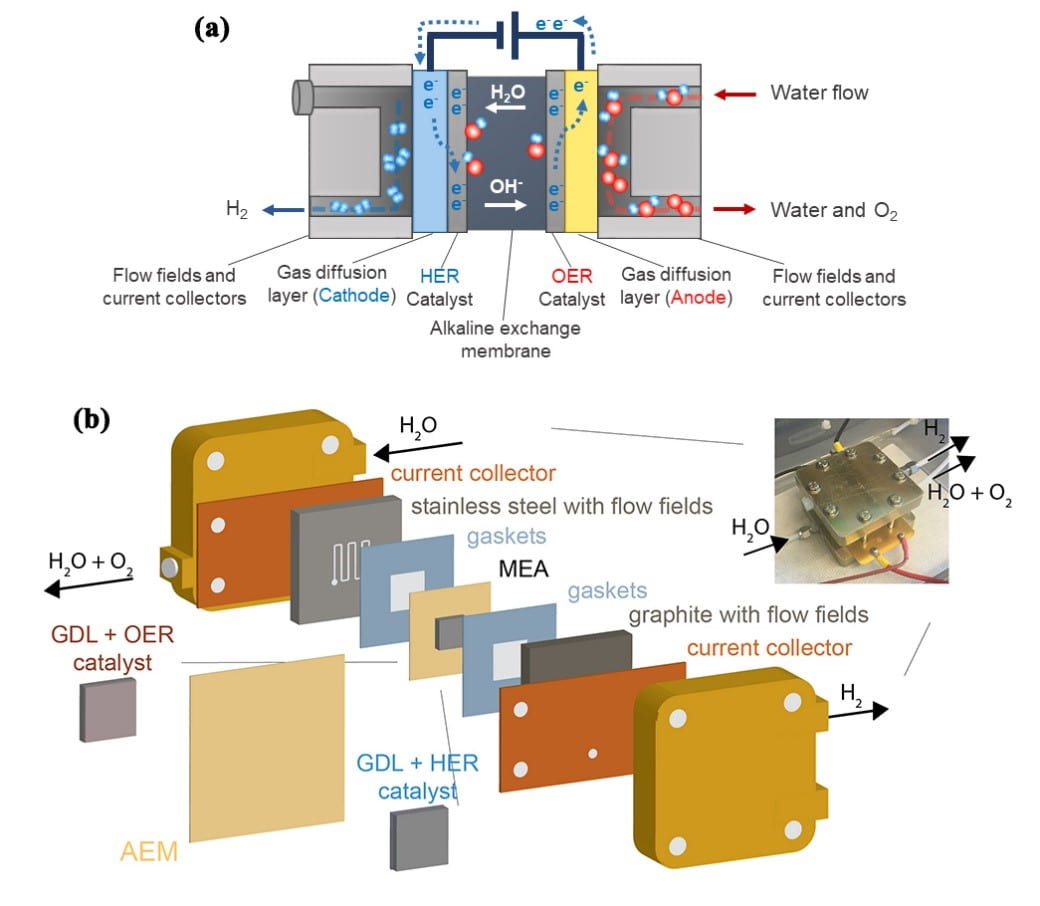
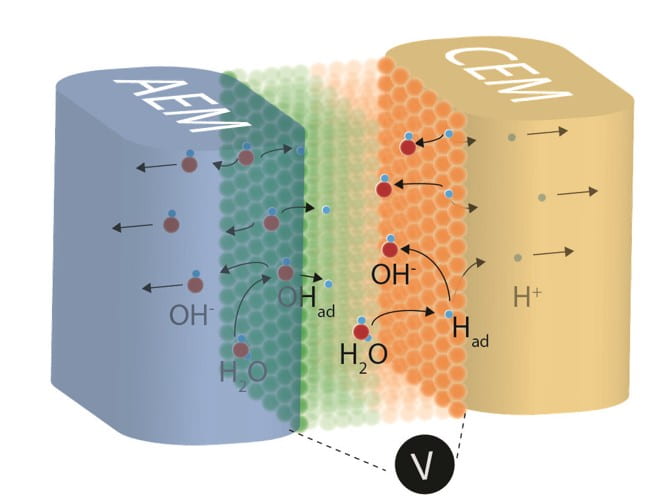
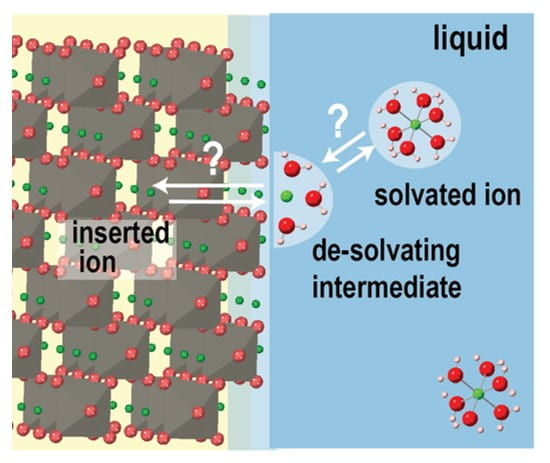
Specific interests include the synthesis and study of heterogeneous electrocatalysts for water oxidation with defined molecular and nanoscale structures, the use of computer simulation and direct electrical measurements to understand semiconductor-electrocatalyst interfaces in photochemistry, the development of alkaline membrane electrolyzers for low-cost scalable hydrogen production, fundamentals and applications of bipolar membranes and electric field driven interfacial ionic reactions like water dissociation and corrosion.
Recent Student PhD Thesis Seminars:
Grace Lindquist – Alkaline Membrane Electrolyzers for Scalable Green Hydrogen
Haokun Chen – Fundamentals of Voltage Driven Water-Dissociation Catalysis in Bipolar Membranes
Video Seminars Delivered by Prof. Boettcher
Water Dissociation Catalysis in Bipolar Membranes and for Electrocatalysis
Towards a Molecular Understanding of Dynamic Fe-based Oxygen Evolution Catalysts
Advanced Bipolar Membranes: Design Principles and Applications in Electrochemical Technology
Towards a Molecular Understanding of Dynamic Fe-based Oxygen Evolution Catalysts
Alkaline Membrane and Bipolar Membrane Electrolyzers for Hydrogen Production
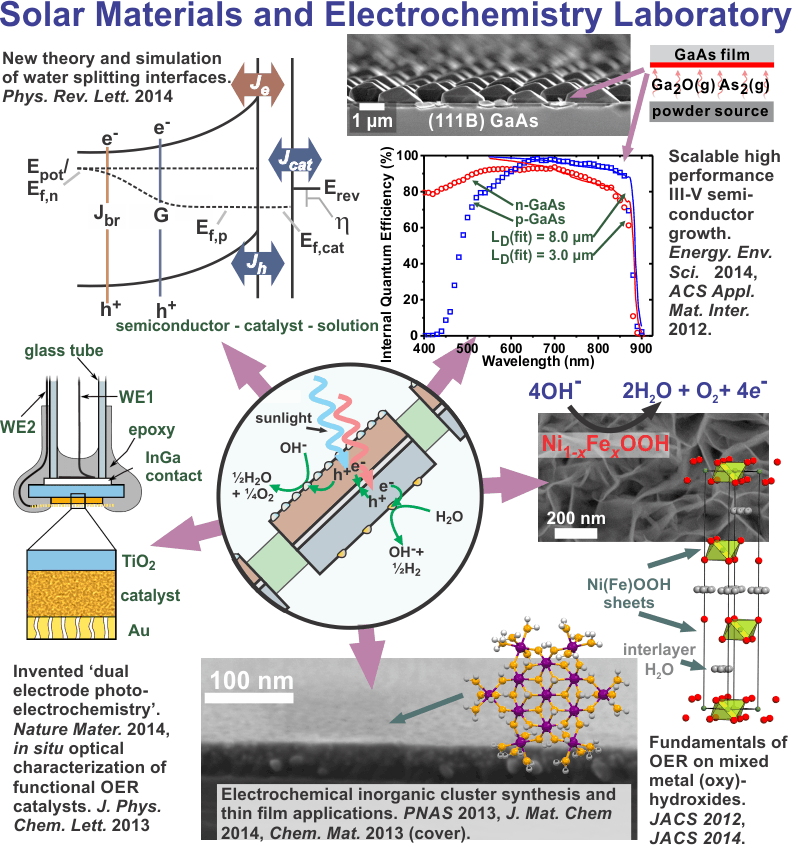
In the past we have also worked closely with groups within the University of Oregon and at Oregon State University through the Center for Sustainable Materials Chemistry. Basic and use-inspired research in the CSMC is focused on developing the fundamental solution chemistry to enable large area precise thin-film deposition from aqueous solution precursors. These materials have applications in electrochromic smart windows, solar cells, water splitting cells, heterogenous catalysis, displays, tunneling electronics, etc.
Motivation
The continued prosperity of our current civilization will require replacing fossil fuels with renewable, sustainable, and carbon-free energy sources. More energy hits the earth in the form of sunlight in one hour than civilization uses in one year. Current photovoltaic (PV) technology has reached “grid-parity” ( < 10 ¢ kW-1 hr-1) and thus drives rapid continued commercial PV growth in the near term.1 Long-term, the critical unsolved challenge of storing massive amounts of energy derived from intermittent renewable sources such as the sun, will limit PVs to providing less than 20-30% of the electricity supply.2 Furthermore, electricity constitutes less than 15% of total energy use worldwide. A replacement for the remainder, i.e. the energy-dense fossil hydrocarbons used in transportation, to heat homes, and to run factories, is also critical.
The development of inexpensive and scalable materials systems that directly convert sunlight into an energy-dense chemical fuel would enable the storage, and hence utilization, of solar energy on a massive scale. The simplest chemical fuel is H2, which could be derived from the photochemical splitting of water and later used in a fuel cell, burned like natural gas, or used as a feedstock to make methanol from CO2. One promising technological vision for a solar water-splitting system is depicted in Figure 1. Such as system utilizes two semiconductors in series.3 These semiconductors absorb light and separate electron-hole pairs. The electrons are shuttled to a catalyst and then used to reduce protons to H2 gas. The positive holes are shuttled to a different catalyst material and used to oxidize H2O to O2 gas and H2. A polymer sheet could be used to both hold the components in place and separate the H2 and O2 gas. Because the excited electrons and holes are used to drive chemical reactions at the semiconductor /catalyst surface, no wires or complicated 3D contacting schemes are needed. These features could lower the costs of water splitting systems compared to traditional photovoltaic systems, while also providing for energy storage in the form of H2. To build such a system and make it efficient, we must develop and understand the fundamental properties of catalysts and semiconductors, as well as the interfaces between them.
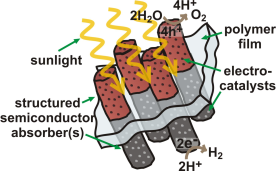
Figure 1. One vision for a solar water-splitting cell that utilizes only sunlight and water as inputs and generates H2 and O2 products. Researchers around the world are working on various fundamental and applied aspects of similar “devices”.
(1) Nemet, G. Learning Curves for Photovoltaics., International Energy Agency, 2010.
(2) Lewis, N. S. Powering the planet. MRS Bulletin 2007, 32, 808-820.
(3) Walter, M.; Warren, E.; McKone, J.; Boettcher, S. W.; Qixi, M.; Santori, L.; Lewis, N. S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446-6473.
Oxygen Evolution Electrocatalysts:
Thin-Film Mixed Metal Oxide and Hydroxide Oxygen Evolution Electrocatalysts: Fundamentals and Applications
The slow kinetics of the oxygen evolution reaction (OER, in basic media: 4OH– → 2H2O + O2 + 4e–) limit the performance of promising large-scale energy technologies based on the photo-driven or electricity-driven production of H2 from water (H2O → H2 + ½O2).1,2 The longstanding challenge is that the active catalysts typically studied are poorly defined, which makes relating structural, compositional, and electronic features to the observed catalysis behavior difficult, especially on heterogeneous surfaces.
We recently demonstrated higher intrinsic OER activity for thin films of Ni-Fe based layered hydroxides/oxyhydroxides [abbreviated as (oxy)hydroxides] than for other known electrocatalysts in alkaline solution and are now working to study these structures in detail.3 Divalent cations (e.g. Mg, Ca, Mn, Fe, Co, Ni) can form hydroxides which are composed of 2D sheets of covalent µ3-hydroxo-bridged metals with the layered-double-hydroxide “brucite” structure (Figure 1). The sheets are held together by non-covalent interactions allowing for the movement of water and ions between the sheets and throughout the structure when it undergoes redox chemistry. The oxidized NiOOH sheets are electronic redox conductors4 and galleries between sheets provide possible pathways for OH– transport.5
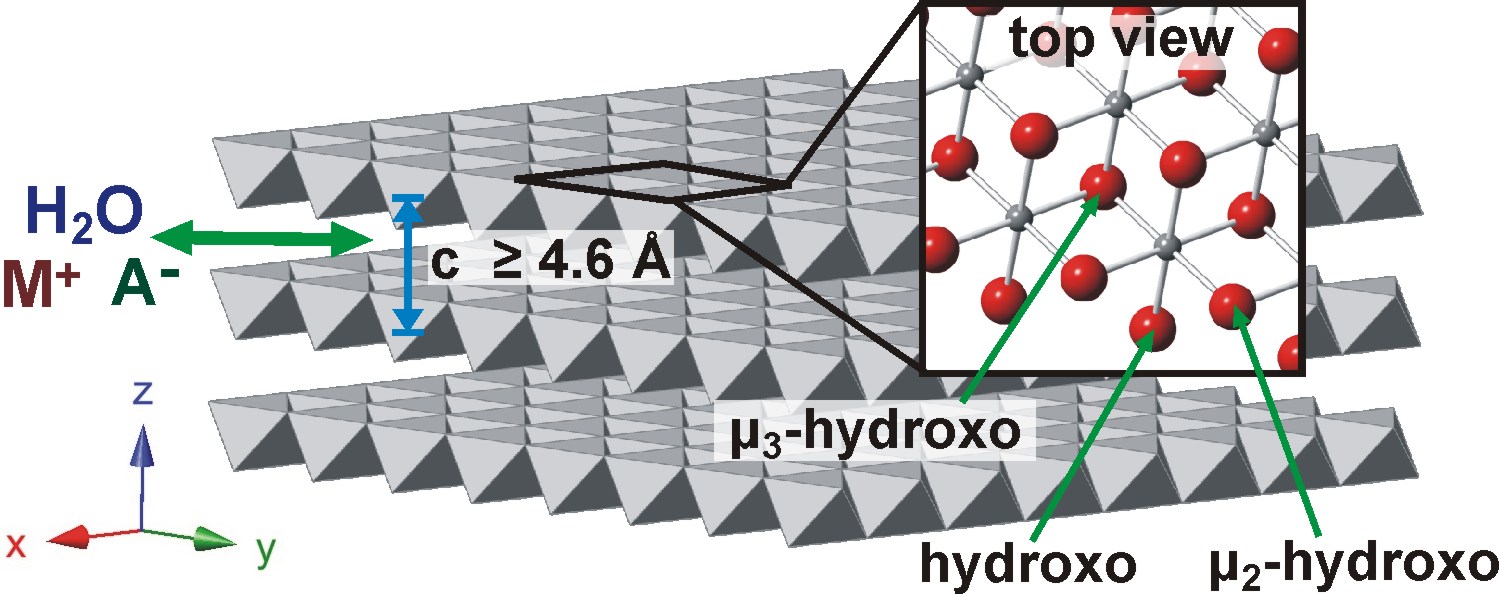
Figure 1. Ni(OH)2/NiOOH structure. The crystalline structure shown above is b-Ni(OH)2. Structures with larger sheet spacing and turbostratic disorder are known as the a-Ni(OH)2. The edge of the sheets are likely terminated with hydroxo and m2-hydroxo groups that are under-coordinated relative to the “bulk” m3-hydroxo bridges. Ions can move in-between the sheets.
We hypothesize that the layered structure of the hydroxide catalysts are especially active because the catalytic sites are sandwiched in-between redox-active transition-metal-cation sheets that modify the energetics of the reaction intermediates differently than is possible on the surface of a dense oxide and that added impurities (e.g. Fe) play a role in this 3D stabilization mechanism. Using synthesis, detailed electrochemical and structural characterization, and theory we are working to address key knowledge gaps surrounding the OER mechanism in these 3D materials and their application in practical devices: How does the sheet spacing influence the electrochemical kinetics and rate determining steps? Is the interior of a Ni hydroxide particle more or less active than the surface? Does the OER mechanism depend on impurities such as Fe in the sheets or do these impurities only affect the electrical or ionic properties? How can these catalysts be electronically and ionically “wired” into advanced alkaline-membrane-based electrolysis systems? Are the catalysts durable in practical operating environments and, if not, can the degradation mechanisms be addressed? To facilitate these efforts we have initiated collaborations with with Profs. Guenter Schneider and Líney Árnadóttir at Oregon State University who are experts in density functional theory calculations as well as with Proton OnSite, the leading manufacturer of commercial high-performance water electrolysis systems.
References:
(1) Cook, T. R.; Dogutan, D. K.; Reece, S. Y.; Surendranath, Y.; Teets, T. S.; Nocera, D. G. Solar Energy Supply and Storage for the Legacy and Non-Legacy Worlds. Chem. Rev. 2010, 110, 6474-6502.
(2) Walter, M.; Warren, E.; McKone, J.; Boettcher, S. W.; Qixi, M.; Santori, L.; Lewis, N. S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446-6473.
(3) Trotochaud, L.; Ranney, J. K.; Williams, K. N.; Boettcher, S. W. Solution-Cast Metal Oxide Thin Film Electrocatalysts for Oxygen Evolution. J. Am. Chem. Soc. 2012, Accepted, DOI 10.1021/ja307507a.
(4) Natan, M. J.; Belanger, D.; Carpenter, M. K.; Wrighton, M. S. pH-sensitive Ni(OH)2-based microelectrochemical transistors. J. Phys. Chem. 1987, 91, 1834-1842.
(5) Tadanaga, K.; Furukawa, Y.; Hayashi, A.; Tatsumisago, M. Direct Ethanol Fuel Cell Using Hydrotalcite Clay as a Hydroxide Ion Conductive Electrolyte. Adv. Mater. 2010, 22, 4401.
Nanoscale Probes of Catalytic and Photoelectrochemical Processes:
Operando potential sensing during electrochemical processes
Heterogeneous electrochemical processes, including (photo)electrochemical water splitting to evolve H2 using electrocatalyst-coated semiconductors, are driven by the accumulation of charge carriers and thus the interfacial electrochemical potential gradients that promote charge transfer. Conventional electrochemical techniques measure/control potentials at a conductive substrate or semiconductor ohmic contact, but cannot isolate processes and electrochemical potentials at the surface during operation.
(Dr.) Michael Nellist in our research group demonstrated that the nanoelectrode tip of an atomic-force-microscope (AFM) cantilever can effectively sense the surface electrochemical potential of operating (photo)electrocatalysts.1 He also studied fundamental aspects and capabilities of the nanoprobes, through a collaboration with Bruker and others (Fig. 1a).2
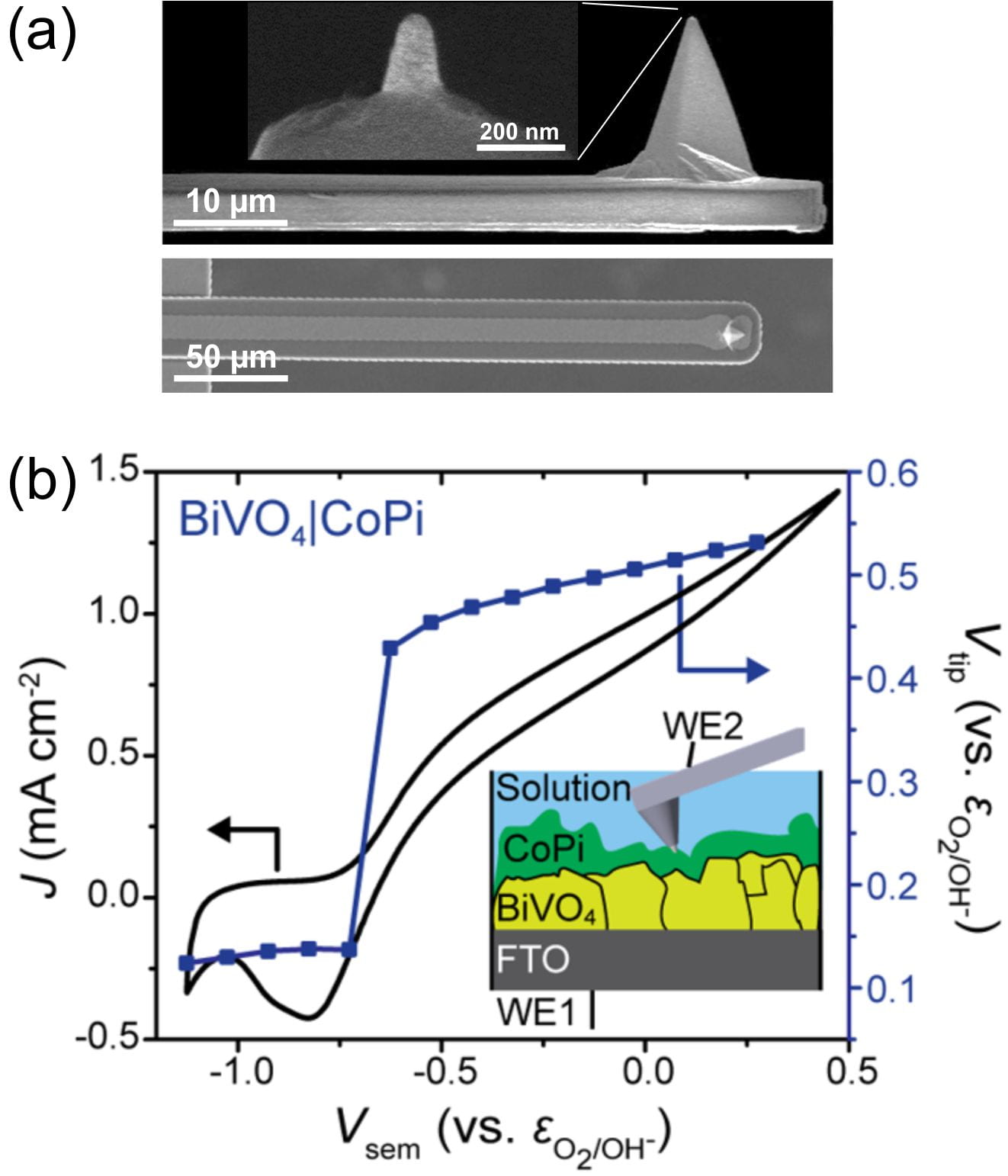
Fig. 1. (a) Electron microscope images of EC-AFM tips used in this study. Only the Pt tip of the conductive probe is exposed. (b) Data collected using PS-EC-AFM shows the catalyst potential (Vtip) as a function of the photocurrent flowing through the structure. The photoelectrode is illuminated from the bottom. The data directly shows that, as the photocurrent onset potential is reached in the current-voltage response, the catalyst is rapidly charged with holes. The potential to which the catalyst is charged is exactly that needed to catalytically drive the oxygen evolution photocurrent.
The potential-sensing electrochemical AFM (PS-EC-AFM) technique allows for measurement of the surface potential and thickness-dependent electronic properties of cobalt (oxy)hydroxide phosphate (CoPi) deposited on illuminated photoanodes such as hematite1 (α-Fe2O3) and bismuth vanadate3 (BiVO4). By comparing the electrochemical potential that the catalyst reaches under illumination on a semiconductor with the potential the catalyst reaches on a metallic substrate passing the same current density, we directly show that metal (oxy)hydroxide layers act as both hole collectors and as oxygen-evolution catalysts (Fig. 2b).4 These results clarify recent conflicting literature results where the (oxy)hydroxide layers were suggested to play a non-catalytic role in enhancing photoelectrochemical performance.5-7
PS-EC-AFM can also be used to spatially resolve charge-separation processes on the nanoscale – critical for understanding how photoelectrochemical assemblies function.8 (Dr.) Forrest Laskowski in my group studied a model system consisting of Ni nanoparticles deposited on n-type Si. This system has been studied extensively as a quasi-stable photoanode for water oxidation under neutral to basic conditions.9 Previous studies have found that the manner in which the Ni is deposited on the Si and how it is electrochemically conditioned dramatically affect the resulting photoelectrode performance. We first measure the Ni nanoparticle / Si interface ex-situ under dry conditions and find that the Ni/n-Si Schottky barrier height (~ 0.6 eV) and photovoltage (~0.3 V) are low and independent of particle size, consistent with traditional bulk measurements. We then measure the nanoparticle/semiconductor junction under photoelectrochemical conditions (Fig. 2a-2c). Remarkably, we find a significantly increased Ni-nanoparticle/n-Si photovoltage (> 500 mV in some cases) and a strong dependence of that photovoltage on the Ni nanoparticle size. The observed size dependence can be analytically described by the so-called “pinch-off” effect (Fig. 3c and 3d).10 In this model, a small-barrier-height nanoscale patch (e.g. the Ni-nanoparticle/Si junction) is surrounded by a larger background barrier height. We discovered, through a series of control measurements, that the background barrier height is set by the oxidized NiOOH that forms in-situ around the Ni nanoparticles (Fig. 2a).

Fig. 2. (a) Schematic showing how the AFM tip measures the photovoltage generated at a single nanoparticle/semiconductor photoelectrochemical junction. (b) Spatially resolved photovoltages at each Ni/n-Si nano-interface. (c) Size-dependent photovoltage fit to pinch-off model. (d) Schematic of major variables in the pinch-off model.
These results not only demonstrate the practical utility of PS-EC-AFM to uncover dynamic nanoscale interfacial properties in situ, but also, fundamentally, provide the first direct experimental observation of the pinch-off effect occurring at the single-nanoparticle level. The results are thus broadly important in understanding and designing nanoscale semiconductor contacts. In the next phase of the work we will apply the new technique to other photocatalytic systems that are poorly understood, for example, co-catalyst modified particulate semiconductors.
- Nellist, M. R., Laskowski, F. A. L., Qiu, J., Hajibabaei, H., Sivula, K., Hamann, T. W. & Boettcher, S. W. Potential-sensing electrochemical atomic force microscopy for in operando analysis of water-splitting catalysts and interfaces. Nature Energy 3, 46-52, (2018).
- Nellist, M. et al. Atomic Force Microscopy with Nanoelectrode Tips for High Resolution Electrochemical, Nanomechanical and Nanoelectrical Imaging. Nanotechnology 28, 095711, (2017).
- Nellist, M. R., Qiu, J., Laskowski, F. A. L., Toma, F. M. & Boettcher, S. W. Potential-Sensing Electrochemical AFM Shows CoPi as a Hole Collector and Oxygen Evolution Catalyst on BiVO4 Water-Splitting Photoanodes. ACS Energy Lett., 2286-2291, (2018).
- Laskowski, F. A. L., Nellist, M. R., Qiu, J. & Boettcher, S. W. Metal Oxide/(oxy)hydroxide Overlayers as Hole Collectors and Oxygen-Evolution Catalysts on Water-Splitting Photoanodes. J. Am. Chem. Soc. 141, 1394-1405, (2019).
- Barroso, M., Cowan, A. J., Pendlebury, S. R., Gratzel, M., Klug, D. R. & Durrant, J. R. The Role of Cobalt Phosphate in Enhancing the Photocatalytic Activity of a-Fe2O3 toward Water Oxidation. J. Am. Chem. Soc. 133, 14868-14871, (2011).
- Ma, Y. M., Kafizas, A., Pendlebury, S. R., Le Formal, F. & Durrant, J. R. Photoinduced Absorption Spectroscopy of CoPi on BiVO4: The Function of CoPi during Water Oxidation. Adv. Funct. Mater. 26, 4951-4960, (2016).
- Zachäus, C., Abdi, F. F., Peter, L. M. & van de Krol, R. Photocurrent of BiVO4 is limited by surface recombination, not surface catalysis. Chem. Sci. 8, 3712-3719, (2017).
- Laskowski, F. A. L., Oener, S. Z., Nellist, M. R., Gordon, A. M., Bain, D. C., Fehrs, J. L. & Boettcher, S. W. Nanoscale catalyst/semiconductor interfaces in photoelectrochemistry. Nat. Mater. Submitted, (2019).
- Kenney, M. J., Gong, M., Li, Y. G., Wu, J. Z., Feng, J., Lanza, M. & Dai, H. J. High-performance silicon photoanodes passivated with ultrathin nickel films for water oxidation. Science 342, 836-840, (2013).
- Tung, R. T. Electron-transport of inhomogeneous schottky barriers. Appl. Phys. Lett. 58, 2821-2823, (1991).
Hydrogen Production via Membrane Water Electrolysis
Earth Abundant Catalysts for Alkaline Exchange Membrane Electrolysis
Anion exchange membrane (AEM) electrolysis is a promising technology to produce hydrogen through the splitting of pure water. In contrast to proton exchange membrane (PEM) technology, which requires precious metal oxide anodes, AEM systems allow for the use of earth-abundant anode catalysts. Here we report a study of first-row transition-metal (oxy)hydroxide/oxide catalyst powders for application in AEM devices and compare physical properties and performance to benchmark IrOx catalysts as well as typical catalysts for alkaline electrolyzers. We show that the catalysts’ oxygen evolution activity measured in alkaline electrolyte using a typical three-electrode cell is a poor indicator of performance in the AEM system. The best OER catalysts in alkaline electrolyte, NiFeOxHy oxyhydroxides, are the worst in AEM electrolysis devices where a solid alkaline electrolyte is used along with a pure water feed. NiCoOx-based catalysts show the best performance in AEM electrolysis. The performance can be further improved by adding Fe species to the particle surface. We attribute the differences in performance in part to differences in the electrical conductivity of the catalyst phases, which are also measured and reported.
Semiconductor-Electrocatalyst Contacts:
Theory, Experiment, and Applications to Solar Water Photoelectrolysis
Semiconductor photoelectrodes coated with electrocatalysts are key components of photoelectrochemical (PEC) energy conversion and storage systems. Despite an intense effort aimed at optimizing these materials, there has been little systematic work focused on the semiconductor-electrocatalyst (SC-EC) interface. The SC-EC interface is important because it responsible for collecting the photoexcited electron-hole pairs generated in the semiconductor. The objectives of this research are to: (1) Understand the rate-/performance-limiting processes at SC-EC interfaces using computer simulations to predict PEC J-E response as a function of materials parameters. (2) Fabricate and study model systems using single-crystal photoelectrodes to verify predictions and determine how the rates of SC-EC electron/hole transfer and the SC-EC interface energetics vary for different catalyst architectures. The catalyst architectures that are studied include dense crystalline oxide films, redox-active ion-permeable oxide/hydroxide films, and monolayers of robust inorganic molecular catalysts. (3) Uncover the interface/catalyst design-principles critical to improving PEC water-splitting photoanodes.
This work focused on SC-EC interfaces will therefore fill key knowledge gaps in the understanding of solar-water-splitting using catalyst-modified semiconductor photoelectrodes. This will enable the design of improved materials systems that are practically relevant because they provide a mechanism to directly convert and store solar energy in the form of hydrogen gas, a renewable chemical fuel.
(project retired) Enabling High-Efficiency GaAs Photovoltaics:
Grown via Vapor Transport from a Solid Source
GaAs is an attractive material for high-efficiency photovoltaics, but its widespread implementation is limited in part by the high cost of metal-organic chemical vapor deposition, which employs toxic and pyrophoric gas-phase precursors. We study the growth of GaAs by close-space vapor transport (CSVT), which uses solid GaAs as a source and water vapor as a transport agent as an alternative, and possibly low-cost, technique for depositing GaAs films. Epitaxial n-GaAs thin films were grown on n+-GaAs substrates while varying the water vapor concentration and the source/substrate temperatures. The photovoltaic properties of the films were evaluated using a non-aqueous photoelectrochemical test cell containing the ferrocene/ferrocenium redox couple.
Remarkably, we found little dependence of the photovoltaic response on the water content used to grow the films suggesting that the incorporation of oxygen-related defects from the water may not limit the materials performance. With 100 mW cm-2 of AM1.5G solar simulation incident on the front surface of the test cell, the best electrodes yielded Jsc = 16-17 mA cm-2, ff = 0.64, Voc > 820 mV, and efficiency ~ 9 %, which was significantly better than high-quality commercial n-GaAs wafers (efficiency ~ 7 %). After correction for reflection losses the spectral response was analyzed to determine the minority carrier diffusion length which for the best films was ~ 1.5 µm compared to ~ 0.7 µm for the commercial wafers. Device-physics modeling shows that solid-state devices could have AM1.5 solar efficiencies as high as ~23% with no additional improvements in materials quality. The films are characterized via photoluminescence lifetime and secondary ion mass spectrometry measurements in order to understand the photovoltaic properties. Current efforts include the fabrication of solid-state CSVT GaAs pn junctions, the growth and characterization of the films on alternative low-cost substrates including Mo and Si, and the introduction of controlled 3D structure for light management.

References:
Ritenour, A. J.; Cramer, R. C.; Levinrad, S.; Boettcher, S. W. Efficient n-GaAs Photoelectrodes Grown by Close-Spaced Vapor Transport from a Solid Source. ACS Appl. Mater. Interfaces 2012, 4, 69-73.
Ritenour, A. J.; Boettcher, S. W. Towards High-Efficiency GaAs Thin-Film Solar Cells Grown via Close Space Vapor Transport from a Solid Source. IEEE PVSC 38, 2012.
High-Performance Solution Processed Inorganic Semiconductors:
For Large-Area Electronics and Energy Conversion Devices
Solution processing is a potentially transformative technology for the large-area deposition of functional inorganic films such as transparent conducting and semiconducting oxides. Despite the great potential, solution processing of dense high-performance inorganic films at low temperature is severely limited by the available inorganic precursor chemistry. Traditionally, inorganic precursors are stabilized in solution by organic ligands and/or large numbers of counter-ions. When organics and non-functional counter-ions are combusted (following deposition) to yield the inorganic film, they leave behind porosity which reduce performance for many applications. Developing new solution precursors that circumvent the use of organic ligands, and ideally, use water as a solvent, are therefore important.
Ideal precursors for solution-processed thin films are defined by the following features: (1) they are highly soluble (~ 1 M or higher) in “green” solvents1 such as water or ethanol so that film thickness can easily be controlled in a single spin-casting step; (2) they contain a minimal number of counterions and no organic ligands that must be later removed and cause porosity, (3) they are difficult to crystallize upon solvent evaporation (i.e., are not simple salts) so that as-deposited films are smooth; and (4) they are stable yet dynamic in solution, poised to crosslink upon mild heating in the solid state to yield dense films. Aqueous all-inorganic nanoscale clusters2-8 have emerged as solution precursors for high-quality, dense, inorganic thin films with electrical properties in some cases equivalent to the best vacuum-deposited films.9-13
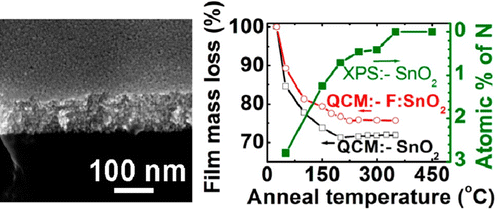
In one example we have developed new aqueous thin-film precursor based on tin(II) hydroxide nitrate clusters.14 This precursor has a number of attractive features, including low-temperature (~100 ºC) inorganic film formation that enables the deposition of conducting F:SnO2 films on flexible plastic films as well as the ability to use F as both a stabilizing ligand for the aqueous precursor and as a functional electronic dopant. The resulting films have electrical performance that is superior to other low-deposition-temperature precursors. In other work we are developing new approaches for semiconducting and conducting films of In-Ga-Zn-O films that have broad applications in macroelectronics.
(1) Capello, C.; Fischer, U.; Hungerbuhler, K. Green Chem 2007, 9, 927.
(2) Donaldson, J. D.; Grimes, S. M.; Johnston, S. R.; Abrahams, I. J Chem Soc Dalton 1995, 2273.
(3) Donaldson, J. D.; Moser, W. J. Chem. Soc. 1961, 1996.
(4) Casey, W. H. Chem Rev 2006, 106, 1.
(5) Son, J. H.; Ohlin, C. A.; Casey, W. H. Dalton T 2012, 41, 12674.
(6) Gatlin, J. T.; Mensinger, Z. L.; Zakharov, L. N.; Macinnes, D.; Johnson, D. W. Inorganic Chemistry 2008, 47, 1267.
(7) Mensinger, Z. L.; Zakharov, L. N.; Johnson, D. W. Inorganic Chemistry 2009, 48, 3505.
(8) Mensinger, Z. L.; Wang, W.; Keszler, D. A.; Johnson, D. W. Chem Soc Rev 2012, 41, 1019.
(9) Mensinger, Z. L.; Gatlin, J. T.; Meyers, S. T.; Zakharov, L. N.; Keszler, D. A.; Johnson, D. W. Angew Chem Int Ed Engl 2008, 47, 9484.
(10) Meyers, S. T.; Anderson, J. T.; Hung, C. M.; Thompson, J.; Wager, J. F.; Keszler, D. A. J Am Chem Soc 2008, 130, 17603.
(11) Jiang, K.; Anderson, J. T.; Hoshino, K.; Li, D.; Wager, J. F.; Keszler, D. A. Chem Mater 2011, 23, 945.
(12) Jiang, K.; Zakutayev, A.; Stowers, J.; Anderson, M. D.; Tate, J.; McIntyre, D. H.; Johnson, D. C.; Keszler, D. A. Solid State Sci 2009, 11, 1692.
(13) Meyers, S. T.; Anderson, J. T.; Hong, D.; Hung, C. M.; Wager, J. F.; Keszler, D. A. Chem Mater 2007, 19, 4023.
(14) Nadarajah, A.; Carnes, M. E.; Kast, M. G.; Johnson, D. W.; Boettcher, S. W. Chem. Mater. ASAP 2013.